
Using Viruses to Cure Sickle Cell Disease?
The methods and ethics of viral vector gene therapy for the treatment of Sickle Cell Disease.
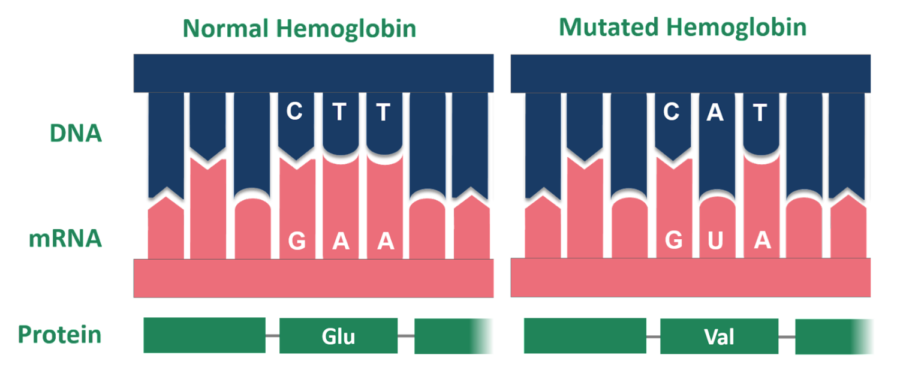
Abnormal hemoglobin has a point mutation, the alteration of a single nucleotide, which changes the amino acid from glutamic acid to valine.
Who would have thought that one day scientists would be using modified versions of pathogenic viruses, like HIV and HPV, to cure rare genetic disorders?
These disorders have always been difficult to treat because the root of the problem lies in the patient’s genetic makeup. One can be prescribed antibiotics to mitigate a bacterial infection, or antiviral drugs to treat a viral infection, but there is no medication that can alter someone’s genes — at least, until recently.
For Sickle Cell Disease (SCD), a rare genetic blood disorder, there may be such a treatment on the horizon: a technology with therapeutic purposes called gene therapy.
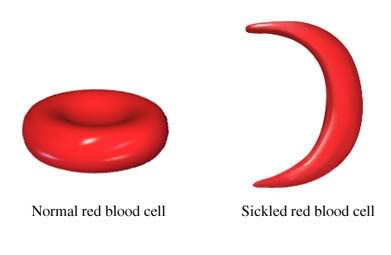
SCD is characterized by the presence of abnormally shaped (crescent-shaped) red blood cells (RBCs). These rigid, crescent-shaped RBCs have the tendency to get stuck in blood vessels and, as a result, block blood flow to certain parts of the body, which can ultimately lead to episodes of severe pain, known as pain crises, or even stroke.
Individuals with SCD typically show signs of the disease at around five or six months of age. These symptoms can include jaundice caused by hemolysis (destruction of RBCs), anemia-induced fatigue (sickled RBCs have a shorter life span which leads to a low RBC count, or anemia), or dactylitis (the painful swelling of hands and feet). Pain crises, high fever, and strokes are some of the more severe symptoms associated with SCD.
These misshapen blood cells are the result of a single point mutation — the addition, deletion, insertion, or alteration of a single nucleotide — to the HBB gene, which encodes the protein beta-globin. Beta-globin is a subunit of hemoglobin, the protein responsible for transporting oxygen throughout the body, and an abnormal form of the subunit will ultimately result in an abnormal hemoglobin, or hemoglobin S (HbS). It is HbS that causes RBCs to adopt the stiff, crescent-shape that has been the defining feature of SCD.
SCD is inherited in an autosomal recessive manner, meaning that only those with two copies of the mutation (one from each parent) will have the disease. Individuals with only one copy, classified as having the sickle cell trait (SCT), will serve as carriers of the disease in that while they most likely will not present symptoms, they are still at risk of passing on the mutation to their children. However, these heterozygous individuals (one copy of the normal gene and one copy of the abnormal gene) actually have a biological advantage over those who have no copies of the mutated gene.
In 1954, South African scientist Dr. Anthony Clifford Allison discovered the link between SCT and malaria resistance. While working as a graduate student in Kenya, Dr. Allison spent his days analyzing blood samples.
“Both sickle cell anemia [the abnormal RBCs] and the malaria parasite can be viewed in the blood using a microscope,” AP Biology and Epidemiology teacher Dr. Erin O’Leary noted. “So Dr. Allison observed that there were less malaria parasites in blood with sickle-shaped red blood cells and more malaria parasites in normal blood.”
This prompted his hypothesis that there was a direct correlation between SCD and malaria resistance, which puts the disease in a unique position: the survival advantage that individuals heterozygous for SCD have outweighs the shorter life expectancy that those homozygous (two copies of the mutation) for SCD have.
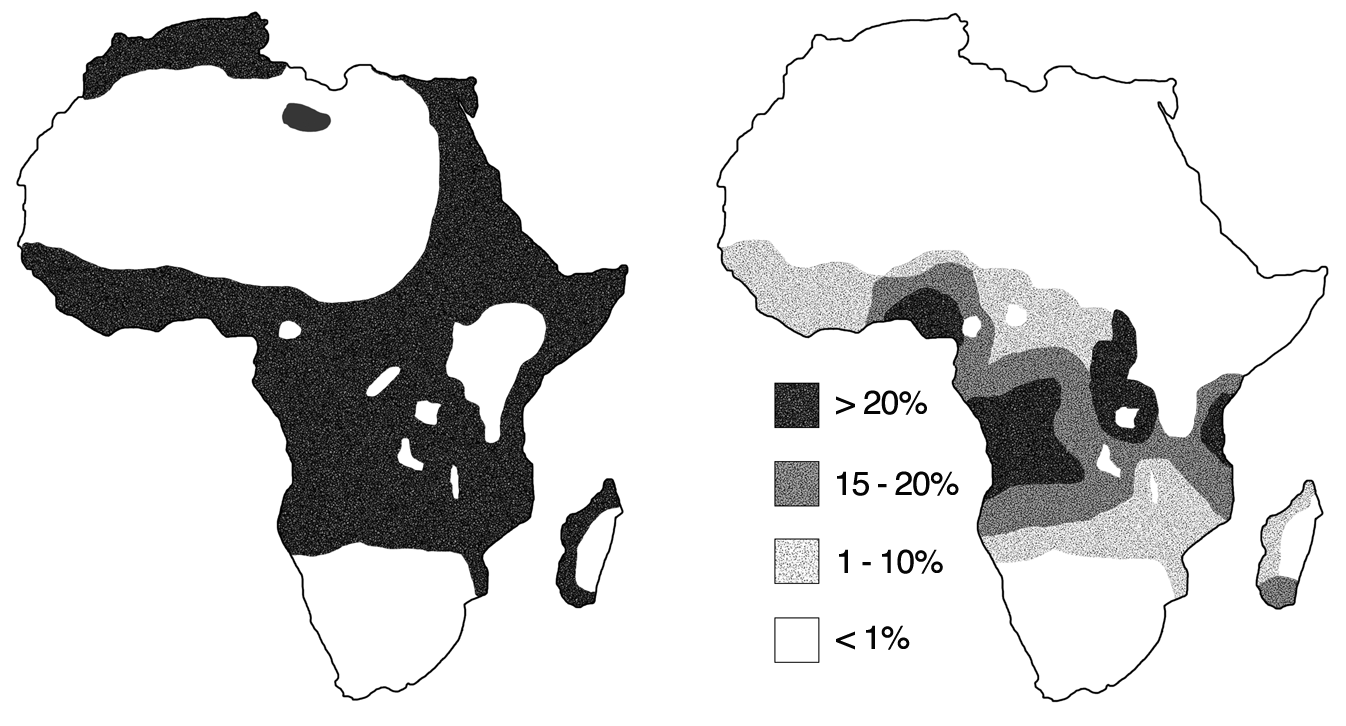
This connection can be observed at large, as areas with higher rates of malaria tend to also have higher rates of SCD — a phenomenon known as the malaria hypothesis. This is particularly true in countries in sub-Saharan Africa, which account for nearly 92 percent of global cases of malaria and 75 percent of births of babies with SCD.
In the United States, an estimated 100,000 Americans, predominantly of African descent, are affected by SCD, and approximately 1 in 13 babies of African descent have SCT. But what happens to individuals with SCT and SCD in areas with a low risk of malaria? SCT no longer serves as a selective advantage, so individuals with SCT are at risk of passing along the genetic mutation to their children.
SCD can be diagnosed in a variety of ways. In order to identify the presence of abnormal hemoglobin, blood samples may be drawn and analyzed using high performance liquid chromatography. In the U.S., all 50 states require newborn screenings, which use blood samples for analysis. There are also prenatal screenings available, such as amniocentesis (using amniotic fluid) or chronic villus sampling (using placental tissue) that would check for the sickle hemoglobin gene, not the presence of abnormal hemoglobin itself.
Treating SCD has historically focused on mitigating the symptoms, rather than ‘curing’ the disease. Medications, such as ADAKVEO and hydroxyurea, have been recommended to manage SCD complications, including pain crises and anemia. Patients with SCD may also receive blood transfusions. Acute transfusions can reduce the effects of severe anemia; RBC transfusions increase the amount of normal RBCs in the body, and ongoing blood transfusions are administered to prevent subsequent strokes.
The only established ‘cure’ for SCD is a blood and bone marrow transplant (hematopoietic stem cell transplant). Bone marrow is where RBC formation, or hematopoiesis, occurs. In this kind of transplant, the cells that are responsible for abnormal RBC formation (abnormal hematopoietic stem cells) are replaced with healthy hematopoietic stem cells. This allows for the production and proliferation of normally shaped RBCs.
Unfortunately, hematopoietic stem cell transplants come at a serious cost, as the patient must undergo chemotherapy and possibly radiation therapy. Furthermore, they can only be performed with someone who has matching bone marrow. These additional requirements make blood and bone marrow transplants not a viable option for many with SCD.
There is, however, a novel treatment that some, including Dr. Julie Kanter, an associate professor in the Division of Hematology and Oncology at the University of Alabama at Birmingham who also runs the Adult Sickle Cell Clinic, believe to yield promising results. This new treatment, called LentiGlobin, relies on a pioneering technology called gene therapy, or more specifically viral vector gene therapy. She told The New Yorker, “We’re not ready to use the word ‘cure’ yet. As best we can tell, it’s an incredibly effective therapy — but we still have to prove that.”
Gene therapy is a new approach to treating diseases, and it focuses on genetically modifying cells in order to produce a therapeutic effect. Viral vector gene therapy, a specific type of gene therapy, uses modified viruses as ‘vectors,’ or vehicles for the delivery of therapeutic genetic material.
Produced by biotechnology company Bluebird Bio, LentiGlobin is one of several gene therapy treatments currently undergoing clinical trials. LentiGlobin involves the ex vivo (out of the body) insertion of a functional version of the HBB gene into a sample of the recipient’s hematopoietic stem cells via a lentiviral vector. This mixture of the vector containing the functional HBB gene and the hematopoietic stem cells is then transplanted back into the recipient, which results in the production of functional hemoglobin.
How exactly does this work? The key is the viral vector. Lentiviruses inject their RNA genome into the cytoplasm of the host cell, whereupon it undergoes reverse transcription via the enzyme reverse transcriptase into a viral DNA genome. In other words, the single-stranded RNA is ‘converted’ into DNA.
Another enzyme called integrase then incorporates the viral DNA genome into the host cell’s genome. Now, the proteins that are encoded by the virus’ genetic material will be produced by the host cell’s own replication machinery.
When it comes to LentiGlobin, scientists can modify the viral genome to include the functional HBB gene instead of viral genes that cause the vector to be pathogenic. So now, when the lentivirus injects its genetic material into the host cell, it will be injecting the HBB gene into the hematopoietic stem cells. As a result, the hematopoietic stem cells will start producing functional hemoglobin.
Bluebird Bio recently finished their phase 1/2 clinical trial for Lentiglobin. From the results published in the New England Journal of Medicine, a total of 35 patients received a LentiGlobin transfusion. 35 patients had normal hemoglobin comprise at least 40 percent of their total hemoglobin, and of those 35 patients, the 25 who could be examined in the follow-up had fewer vaso-occlusive events or pain crises (a 99 percent reduction).
As Bluebird Bio is currently recruiting participants for their phase 3 study, it is important to acknowledge the significant potential of these results. Instead of relying on treatments that help manage symptoms, “We’re starting to talk about curative-intent therapies at visits within the first year of life,” said Anjulika Chawla, a pediatric hematologist oncologist, to The New Yorker.
A shift from treatment plans that only focus on the effects of the disease to treatment plans that target the root of the disease itself offers a more positive prognosis for patients who usually have an average life expectancy of 42 to 47 years. This also presents a unique offer: a one-time treatment without the need for daily disease management.
While this all sounds incredibly promising, Dr. O’Leary cautioned, “Scientists are still only just starting to use gene therapy, and long term studies are needed to determine any possible side effects. I think there is the potential for gene therapy to lower the prevalence of the disease, but it will take more time and studies for this to happen.”
Similarly, questions about the accessibility of gene therapies for SCD linger. The idea of putting a price or receiving compensation for something as necessary to life as a treatment for a disease is controversial. Jonas Salk, an American researcher behind the development of one of the first successful polio vaccines, famously did not patent his own creation — though there is still some argument as to whether this was entirely altruistic or whether his patent would have been rejected anyway for failing to meet the novelty requirements. This debate has also extended into U.S. politics, as House Democrats recently passed The Affordable Insulin Now Act, which would cap the cost of insulin.
Mandeep Kaur ’22, Vice President of the Bronx Science Bioethics Club, which meets weekly to discuss various ethical challenges that arise as a result of scientific progress, chimed in. “If gene therapy becomes widespread, I think there will be a lot of tension as to the financial implications of its availability.”
Bluebird Bio has already come under fire for its costly treatments. The company’s gene therapy (Zynteglo) for the rare genetic disorder beta thalassemia has a $1.8 million price tag, making it the second most expensive drug in the world. There is indication that LentiGlobin, a similar form of gene therapy to Zynteglo, would bear a comparable cost. Bluebird Bio has argued that its treatments are cost-effective, as patients will no longer have to receive frequent blood transfusions which can total up to $400,000 per patient decade (including the cost of RBC transfusion and chelation therapy to remove excess iron).
Nevertheless, Kaur remains skeptical. “Such treatments will not be as readily available to lower-income patients as they will be to wealthy patients, which simply worsens the U.S.’s preexisting income inequality. How can something be touted as a potential cure if not everyone will have access?”
It is not simply LentiGlobin’s price that has some concerned. There are also ethical implications that arise with the use of gene therapy, which often draws comparisons to genetic engineering.
“Genetic engineering and gene therapy are conflated in that gene therapy essentially replaces or modifies an aspect of someone’s genetic makeup in order to improve function, while genetic engineering modifies the genes in order to either introduce new characteristics or enhance what already exists,” explains Kaur. “The main difference between the two lies in their purpose, given that gene therapy is usually used as a method of treatment, but genetic engineering can have more cosmetic than corrective implications.”
That difference is where the controversy lies. “Most people generally agree that gene therapy is more acceptable and beneficial because it is used to improve the health of its recipients,” said Kaur. “Genetic engineering’s potential cosmetic uses, on the other hand, invoke ethical considerations for its potential misuses that make it harder to rationalize.”
As with any sort of new development, the never-ending debate over its merits and drawbacks rears its head. Nevertheless, if its issues in accessibility are addressed, gene therapy, with its myriad of potential applications, remains a technology that represents an exciting new step forward for medicine.
“Such treatments will not be as readily available to lower-income patients as they will be to wealthy patients, which simply worsens the U.S.’s preexisting income inequality. How can something be touted as a potential cure if not everyone will have access?” said Bioethics Club Vice President Mandeep Kaur.
Meriel Crowley-Wang is an Editorial Editor for ‘The Science Survey.' She enjoys the logical way in which journalistic writing presents itself to the...